熱門資訊> 正文
國產新葯赴美上市審評,FDA的「公開課」講了什麼?
2022-02-23 21:32
本文來自微信公眾號:同寫意(ID:tongxieyi),作者:萬里(啟德醫藥高級副總裁和國際註冊事務部負責人),審稿:應嘉、夏毅、洪筱玲、黃桂林、杜新,原文標題:《FDA的這堂公開課到底講了什麼?——信達/禮來ODAC會議的啟示》,頭圖來自:IC photo
美國時間2月10號,備受矚目的美國食物藥品監督管理局(FDA)為信達/禮來PD-1抗體信迪利單抗(sintilimab)與化療聯用作為非小細胞肺癌一線療法的新葯上市申請(BLA)的腫瘤藥物專家委員會議(ODAC會議)如期召開。
經過5個多小時的討論並投票表決,腫瘤藥物專家委員會最終以14票贊成對1票反對的壓倒性投票支持FDA的提議,要求信迪利單抗在獲批前需要補做新的臨牀試驗。筆者雖然對於這次ODAC投票結果感到遺憾,但是認為信達作為中國生物製藥公司中的領頭羊勇於出海,成為第一個吃螃蟹與FDA開ODAC會議的中國創新葯企業,這種挑戰自我的精神還是值得肯定的。
FDA通過這次的ODAC會議向所有人尤其是中國的生物製藥公司明確了對於來自中國的單一國家臨牀數據到美國申請新葯上市的准入要求,從ODAC會議的結果也可以看出來中國的生物製藥公司在對於國際監管機構的註冊要求和政策變化的理解,全球註冊策略的制定,臨牀設計和執行等方面與FDA的要求還是有不小的差距。
對於信達生物而言,這次的ODAC會議結果無疑讓信迪利單抗美國上市和中國PD-1產品出海暫時受挫,但對於國內眾多致力於創新葯研發的公司來説,卻是由FDA免費上了一次寶貴的公開課。而對每個有創新葯出海的國際化雄心,並希望早日構築國際競爭優勢的中國生物醫藥公司來説,熟悉FDA等國際監管機構監管規則和要求,懂得如何與FDA溝通,瞭解如何設計和執行MRCT臨牀試驗,更是迫切需要的必修課。
那麼FDA在ODAC會議這堂公開課上到底講了什麼?而準備出海的中國藥企又可以從這堂公開課上學到什麼?筆者根據多年的國際註冊經驗,對於這次ODAC的會議內容和FDA的審評要求進行深入解讀,希望能爲準備出海的中國藥企的提供一些有益的幫助。

一、新葯研發需要從早制定全球註冊策略,並且與時俱進、貫穿始終
一個新葯的開發,平均需要超過10億美金規模的資金投入以及10到15年的時間投入,才能在新葯全球上市后得到商業回報。新葯開發的臨牀周期如此長,花費如此高,所涉及的因素又如此多,新葯研發如何確保臨牀開發方向的最優化?如何做出科學合理的判斷並決定繼續還是終止臨牀試驗?怎麼確保臨牀數據滿足監管機構的上市要求?
一般來説,爲了提高新葯研發的成功率,任何一個新葯的研發一旦進入臨牀階段,就需要制定一個非常全面合理的臨牀開發計劃(CDP,Clinical Development Plan)和相應的全球註冊策略(GRS,Global Regulatory Strategy)。
臨牀開發計劃是一個新葯臨牀開發的整體計劃,而全球註冊策略則是支持臨牀開發,並在全球成功獲批上市,並達成目標產品概況(TPP,target product profile)的藍圖。
全球註冊策略一般按照以終為始的方法,根據產品的最終臨牀需求(比如產品説明書product label)和監管機構的上市要求來反推需要的臨牀數據,根據不同地區監管差異和特點來確定每個地區的最佳申報途徑,由一個包括臨牀、註冊、 CMC、非臨牀、市場、和醫學事務的跨領域團隊(cross-functional team)共同完成。
歐美的藥企傳統上會先考慮歐美日的上市要求來制定這在這三個主要市場上市的註冊策略,隨着中國成為世界第二大藥品市場並在2017年加入ICH,過去幾年中國監管政策與國際逐漸接軌,新葯臨牀申請(IND)審評時間也縮短到了大概三個月,使得越來越多的歐美公司在一開始制定國際註冊策略時,就會考慮在中國做橋接試驗或者MRCT臨牀試驗以加快在中國的上市速度。
尤其是在2017年后,ICH推出了E17文件來指導如何使用MRCTs的平行全球註冊策略來替代之前的E5文件中用橋接試驗來外推其他地區臨牀試驗結果的策略。國際多中心試驗可以更有效的提高臨牀開發速度,避免臨牀資源的浪費,明顯優於單一國家臨牀加上不同地區橋接試驗的做法。
而對於把目標定位在國際新葯研發和海外產品上市的中國藥企,從一開始制定註冊策略就應該考慮中國以外地區,尤其世界第一大藥品市場——美國的上市要求,通過MRCT來加快在中國以及海外的上市速度。
值得一提的是,新葯產品一旦獲得了美國FDA的上市批准,也意味着得到了世界上最高水平的監管機構的認證,就可以免去新的臨牀試驗而通過藥品證書CPP(Certificate of Pharmaceutical Product)申報的方式在很多接受FDA藥品證書的其他國家和地區(比如亞洲,拉丁美洲,南美洲,中東和非洲)獲批上市,也就是説FDA批准不僅是打開美國市場的鑰匙,也是打開世界市場的鑰匙。
在ODAC會議上,信達/禮來提到ORIENT-11一開始只是計劃在中國上市批准,所以臨牀設計也是按照中國的註冊要求來做的,但是在2020年初獲得ORIENT-11臨牀試驗的中期分析數據后,同時看到2019年FDA腫瘤學卓越中心主任Richard Pazdur博士在AACR年會上表示完全來自中國數據的藥物只要質量足夠高就可以到美國申請上市,才決定向FDA提交了ORIENT-11臨牀試驗的結果並申請上市批准。
從註冊的角度來看,信迪利單抗的開發本來只是考慮了中國註冊要求可以理解,但是缺乏一個完整全面的全球註冊策略難免給產品的出海之路帶來很多一開始預料不到的困難,也造成了ORIENT-11的臨牀設計和結果與FDA的註冊要求存在許多差距。
除了熟悉FDA的註冊要求,中國藥企也需要密切關注每個國家尤其是美國監管政策的變化,腫瘤藥的開發競爭激烈,臨牀進展更是日新月異,所以任何一個新葯的臨牀開發計劃和全球註冊策略都需要與時俱進,隨時瞭解競品開發和監管政策的改變,從而做出相應的變化。
Richard Pazdur博士也在ODAC會議的最后決議之前特意發言解釋了説他在AACR年會上發言之后改變想法的原因,提到了很多因素都有了改變,所以新葯的開發策略應該以與FDA的充分溝通爲準,而不應該僅憑他在某個公開場合的講話。
誠如Richard Pazdur博士所説,美國的監管政策最近幾年確實有很多新的變化,第一是FDA對於PD-1產品的批准正在收緊。FDA過去7年已經批准了7個PD-1/PD-L1的抗體藥品,超過85種的適應症,其中很多是以替代終點批准的加速上市。所以FDA在去年四月特地召開ODAC會議重新評估是否繼續批准未在驗證性試驗中顯示出臨牀療效的三大PD-(L)1的六個加速批准適應症,並在去年共撤回了7個腫瘤藥的加速批准適應症,還建議Agenus撤回其PD-1單抗balstilimab用於化療后疾病進展的復發或轉移性宮頸癌患者的上市申請。
今年一月賽諾菲(Sanofi)和再生元(Regeneron)也由於未能與FDA就上市后研究達成一致而撤回了PD-1抗體Libtayo二線治療晚期宮頸癌患者的上市申請(sBLA)。同時在非腫瘤領域,2021年給予Biogen公司治療阿爾茨海默症的藥物Aduhelm上市批准就引起了很大的爭議,也讓FDA受到了公眾很大的壓力,覺得其對於新葯的上市批准尺度太松。
第二是FDA確實在大力倡導Project Equity,並在2020年推出了提高臨牀試驗人羣多樣性的指導原則,希望把黑人患者在內的美國少數族裔包括到臨牀研究中,FDA在過去兩年中幾乎所有新的NDA/BLA批准中都要求上市后臨牀研究PMC中納入黑人患者。
考慮到禮來作為一家老牌的美國藥企,從2015年就與信達合作,直到2020年簽訂協議買下信迪利單抗的海外權益,所以理論上禮來應該憑藉其對於美國監管政策的熟悉給以信達指導,對於這個結果禮來顯然難辭其咎。
不出意料的是FDA在ODAC會議上把矛頭主要對準了信達生物在美國的合作伙伴禮來,Richard Pazdur博士在會議上質問禮來為什麼在承諾增加試驗多樣性的同時,用這樣一個完全來自中國的單一國家臨牀數據來提交上市申請。甚至在討論中提到雙方溝通時間的陳述中出現了一些充滿火藥味的語句,禮來聲稱在提交BLA之前雙方溝通過三次。
FDA腫瘤二科主任Harpreet Singh博士則表示FDA一直到2020年4月的pre-IND會議中才知道信達做了ORIENT-11臨牀試驗並準備申報BLA,並在與信達的溝通中提出了非常多的擔憂,而禮來意外的公佈了一頁2020年8月與FDA的Pre-BLA的會議紀要,也讓FDA負責人非常惱火,甚至提出要向公眾公開所有的溝通文件,禮來只有在會上進行道歉,表示無意曲解與FDA的溝通和誤導ODAC委員會。
一般來説,選擇在ODAC會議上與FDA這樣針鋒相對的激烈討論需要非常慎重,因為FDA大多是在產品的獲批存在重大爭議或者有很大不確定性的時候纔會召開ODAC會議。有經驗的藥企還是需要從臨牀開始階段就與FDA保持良好的溝通和信譽「credibility」,讓FDA對你的公司,產品和數據都有信心,在有爭議的時候儘量通過良好溝通來解決,而不要選擇ODAC會議這樣的方式。
由於ORIENT-11試驗本來就是按照中國上市的要求進行設計的,沒有考慮到美國上市的要求,所以出現很多的硬傷也並不奇怪。如果只是一兩個問題,那麼還是有可能與FDA通過協商PMC/PMR來解決,因為FDA的審評原則是看證據鏈總體性「totality of evidence」,可是太多的硬傷就很難通過溝通來彌補。
相信如果信達/禮來與FDA儘早進行溝通,獲得FDA對於註冊研究臨牀設計的反饋及時調整臨牀方案修補硬傷,而不是等到藥物提出上市申請的最后一刻才進行,那麼這次ODAC會議的結局也許會不一樣。
而在與監管機構溝通的時候還需要考慮到中美兩國監管機構的風格的不同,CDE像一個教練,會直接指導企業應該在研發中做哪些工作,而FDA更像一個裁判,不會直接指導企業怎麼做,而是希望企業提出自己的開發想法后給予具體的判斷意見。所以企業在設計臨牀試驗尤其是申報上市關鍵臨牀的時候就需要充分與FDA溝通,獲得FDA的認可后再開始臨牀試驗,這樣可以幫助企業少走很多不必要的彎路。
而在與FDA在新葯上市申請中的溝通更是一項複雜而繁瑣的系統工程,需要通過pre-NDA/BLA meeting,orientation meeting,mid-cycle communication, late-cycle meeting等一系列審評會議與FDA保持良好溝通,及時就FDA的問題和發補完成答覆,避免新葯審批的延誤,確保早日獲批上市。
總而言之,新葯研發需要從早制定全球註冊策略,並且與時俱進貫穿始終,同時與FDA建立起暢通的溝通交流,讓FDA對所有的新葯研發和臨牀試驗進展保持知情,這將大大提高新葯臨牀和上市獲批的成功率。
二、臨牀試驗設計是一項需要跨部門合作的工作
衆所周知,臨牀試驗的方案設計是一項需要跨部門合作的工作,一個高質量的臨牀試驗設計離不開醫學,臨牀運營,臨牀藥理,數據管理、生物統計,臨牀安全,和註冊等各個部門的緊密配合和綜合考慮。
ORIENT-11試驗的多個硬傷其實是在試驗設計就有的問題,當初臨牀設計可能只是簡單地照搬了Keynote的試驗設計,滿足了當時註冊策略中的最低要求,沒有讓有經驗的統計和註冊來進行把關。
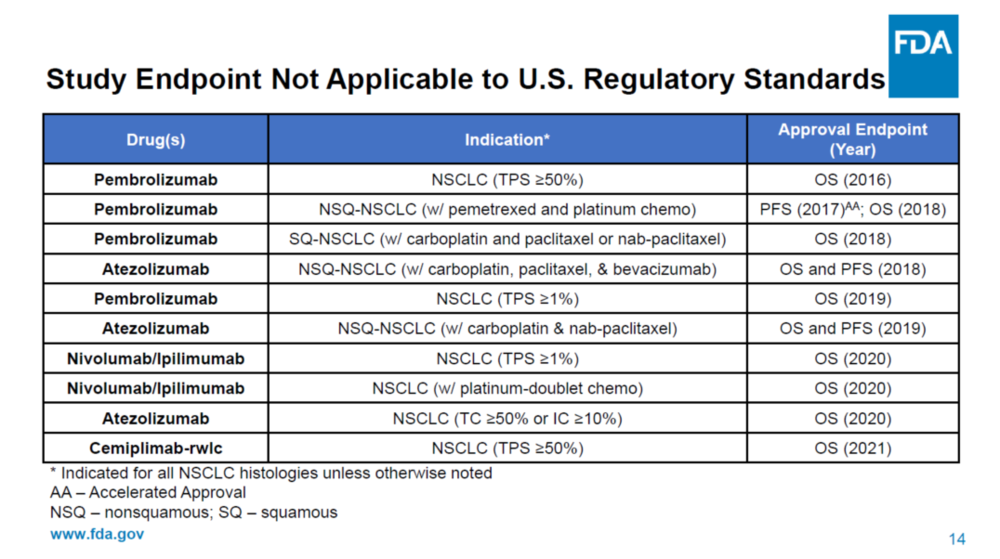
FDA在ODAC會議上沒有質疑ORIENT-11試驗的有效性和安全性,但是提出導致其數據無法推廣到美國人羣的多個缺陷,主要包括:主要終點採用了PFS而不是OS,使用了化療而不是已經上市的免疫療法做對照組,所有入組病人都是中國人而沒有歐美人種的數據,知情同意書沒有按照美國要求及時更新,以及可能存在缺乏數據完整性的問題。
如果信達在這個臨牀試驗啟動或者進行中與FDA溝通,FDA會要求把對照組改成已經上市的Keytruda加上化療,因為在入組第一個患者前這個組合在美國已經批准用於一線肺癌,也會建議用OS而不是PFS作為終點。信達答覆中提到ORIENT-11本來只是計劃在中國上市,所以整個設計當時只是考慮滿足中國的註冊要求,而中國CDE的指導原則《晚期非小細胞肺癌臨牀試驗終點技術指導原則》中明確提出可以接受單獨PFS作為晚期NSCLC註冊研究的主要終點。
迄今為止,FDA對轉移性非小細胞肺癌一線免疫治療(IO,Immuno-Oncology therapy)產品的所有上市完全批准(full approval)都是基於總體生存OS的統計學上的改善。免疫治療相對於靶向治療和化療的優勢主要體現在OS生存期的延長,所以TKI藥物的批准是可以用PFS作為終點的,但是所有PD-1藥物的完全批准都要求用OS作為終點。
Keytruda的Keynote-189三期臨牀一開始也是用PFS做主要終點,但是后來加上了OS作為主要終點,在試驗成功后獲得了非小細胞肺癌一線治療的完全批准上市。
ODAC會議上也討論到在沒有預先設定嚴格的OS分析的情況下,怎麼評價ORIENT-11的OS數據?信達/禮來解釋説,雖然ORIENT-11沒有事先定義嚴格的OS假設檢驗,但是在PFS中期分析之前已經有了OS觀察的計劃,然后也有通過方案修訂將最終時間定在最后一個患者被隨機入組后大約兩年,並在會議上展示ORIENT-11的post-hoc OS分析結果是有效而穩健的。
但是信達/禮來的這個解釋顯然非常無力,如果沒有預先設定的OS分析,包括分配alpha值、多重性校正以及定義OS目標事件數等,而只是做post-hoc分析得出的OS數據只是描述性的,缺乏統計的嚴謹性。就像沒有在開槍之前就畫好靶子,無論之后打的有多準都無法説明射擊的準確性。
FDA也明確迴應這些分析只能支持假設生成hypothesis generating,不能用於假設驗證hypothesis testing。如果對於FDA的審評特點有所瞭解,就會知道FDA對於關鍵臨牀試驗的設計和試驗執行過程中數據完整性和統計嚴謹性的重視。
FDA有一個非常大的統計部門負責統計審評,在關鍵臨牀試驗之前會對試驗設計提出指導意見,在新葯申請時要求申請人提供所有的臨牀試驗數據,由FDA的統計師來獨立計算,確認所有的結果。
筆者認為如果信達在試驗設計初期把OS作為一個關鍵次要終點(key secondary endpoint),採用gatekeeping守門法的方式,在PFS分析成功之后對OS做分析,並不會消耗alpha值影響試驗成功,也不增加整個試驗的複雜度,反而可以額外獲得嚴格意義上的OS結果。
值得一提的是,新葯上市雖然是BLA的主要目的,但是更重要的是新葯最終能夠成功launch product讓病人獲益並獲得商業上的成功,與FDA就產品説明書Product Label的討論是重中之重,因為美國的醫藥銷售和醫生開藥的時候最重視的就是Label上的有效性和安全性説明。
Label上的每一條聲明都是需要用臨牀數據來支持,並在臨牀試驗設計的時候就考慮到這些需求,所以一般有經驗的製藥公司會在設計關鍵2/3期臨牀試驗的時候,即使只是用OS作為次要終點(secondary endpoint),也會給出統計假設並事先分配alpha值,因為這樣得到的OS的數據就可以放到Label上,在市場上已經有很多競品的情況下更有機會被醫生青睞,從而促進病人使用和產品銷售。
三、臨牀執行的細節決定試驗的質量
除了與FDA溝通和臨牀設計等問題,FDA對於 ORIENT-11試驗在試驗人羣選擇,倫理的合規性,和臨牀數據的完整性也提出了質疑。
FDA稱這個ORIENT-11用於支持審批的三期臨牀試驗完全在中國進行,而不是全球多中心臨牀試驗,因此並未反映美國肺癌患者的種族和民族多樣性,不再符合Project Equity的精神,接受此類研究與行業範圍內對臨牀試驗公平性、多樣性的承諾存在衝突。
但是這並不表示,FDA完全不接受海外臨牀數據包括中國數據,其實FDA對於海外數據的接受政策已經在21 CFR 312.120and314.106中給出了非常詳細的規定,需要達到三個要求:
1. 國外數據適用對美國人口和醫療實踐;
2. 研究已經由具有公認能力的臨牀研究人員進行;
3. FDA可以通過現場檢查或其他適當方法驗證臨牀數據的可靠性。
之前也不乏用海外數據獲批上市的產品,比如2019年百濟神州的澤布替尼Brukinsa基於在中國的關鍵性二期BGB-3111-206試驗數據獲得FDA加速批准上市用於復發難治性套細胞淋巴瘤的治療,2021年日本第一三共製藥(Daiichi Sankyo)靶向HER2的抗體藥物偶聯物(ADC)藥物Enhertu基於在日本和韓國的關鍵二期DESTINY-Gastric 01試驗的結果獲得FDA批准用於治療已接受過曲妥珠單抗治療的局部晚期或轉移性HER2陽性胃癌或胃食管交界腺癌病人。
當然這兩個適應症在美國都是罕見病,而這兩個產品也都顯示了非常顯著的臨牀療效,滿足了FDA對於海外數據的接受要求,所以FDA給予相應的監管靈活性批准上市。
FDA對於信迪利單抗試驗的對照組選擇也表現出極大不滿,因為這涉及到倫理審查問題。FDA在討論中提到如果信達提前與他們溝通的話,FDA肯定會要求把對照組改成K藥+化療做非劣效研究來滿足要求,因為在入組第一個患者前這個組合在美國已經批准用於一線肺癌。
Harpreet Singh博士對禮來發出質疑:「禮來對剝奪患者延長總體生存期的治療的化療組感到滿意嗎?禮來進行了多少次試驗,剝奪了患者獲得已知生存優勢的治療方法?」Richard Pazdur博士也表示:「我對這個問題感到非常不舒服,因為已知療法可以改善一年以上的平均生存率,而患者卻沒有得到它。我希望委員會成員就這部分進行一些討論。」
筆者認為FDA在這個問題上有點苛求,考慮到ORIENT-11臨牀試驗啟動時K藥並未在中國獲批上市,採用化療作為對照組也可以理解。
FDA還強調了ORIENT-11試驗在知情同意程序方面的疏忽,尤其是在2019年3月Keytruda獲得中國藥監局批准上市聯合化療用於一線非小細胞肺癌之后,知情同意書在試驗期間沒有按照要求更新,讓病人獲知有新的免疫治療產品獲批上市。知情同意程序沒有讓病人獲得充分地知情權,也不符合美國臨牀實踐。ODAC會議上專家也認為,ORIENT-11試驗未能達到FDA的知情同意標準,因為它沒有明確列出經批准的療法或參與替代研究的治療方法。
美國國家癌症研究所臨牀主任Ravi Madan教授表示:「雖然數據完整性在臨牀研究中至關重要,但倫理方面的完整性更為重要。」信達生物和禮來也在會議上表示已經認識到在知情同意程序上的不足,並已經更新了內部的知情同意流程,確保在病人入組時提供完整的信息,讓病人明確瞭解當時已有的治療方法,可以自己判斷是否同意入組臨牀研究。
FDA在ODAC會議上指出在中國研究者參加國際多中心的臨牀試驗經驗有限,還直接引用中國食品藥品監督管理局2016年報告,稱80%的中國臨牀研究存在造假或不合格。事實上自2015年7.22臨牀數據覈查后,臨牀試驗的真實性、規範性和完整性一直是中國新葯研發重點監管對象,中國新葯臨牀研究已與國際接軌,數據不真實已幾乎不存在。FDA對ORIENT-11試驗的48個臨牀中心中的兩個中心進行GCP覈查也證實了沒有發現任何數據完整性的問題。
筆者認為,在這個問題上,FDA援引6年前的報告的做法顯然是對於中國臨牀有些刻板印象之嫌,信達的迴應非常有力,澄清了這些年來中國臨牀研究在試驗質量、數據完整性和監管都有所提高的事實,讓大家瞭解到中國臨牀研究現在是能達到國際標準的。
但是FDA對於ORIENT-11試驗在人羣選擇,倫理的合規,和臨牀數據的真實性的質疑提醒了中國企業將來在做臨牀研究時,一定要更加註重臨牀執行的細節,一開始就按照國際標準做好臨牀設計和執行,以保障臨牀試驗的質量和GCP合規性。
四、中國PD-1產品美國上市還有機會嗎?
FDA非常罕見地在這次的ODAC會議之前在新英格蘭醫學雜誌(NEJM)和柳葉刀(Lancet)等不同的專業期刊和媒體上發表了幾篇文章,指出美國和世界範圍內PD-1/PD-L1抗體的過度開發的狀況,雖然FDA已經批准了7個PD-1/PD-L1抗體,但是FDA仍然收到了大約有25個來自中國公司PD-1/PD-L1抗體的新葯上市申請,而且這些申請幾乎都是隻有中國數據。
可以説FDA在這次ODAC會議之前已經對於信迪利單抗的上市申請定了基調,但是FDA顯然還是希望通過這次的ODAC會議向所有人尤其是中國的生物製藥公司,明確對於來自中國的臨牀數據到美國申請新葯上市的准入要求。
雖然説ODAC會議投票意見不具有對FDA決策的約束力,但是考慮到FDA在這次ODAC會議之前和會議上的表態,基本上已經可以推斷,等3月22號PDUFA日到期時,FDA大概率會給出一封拒絕批准上市的完整迴應函(CRL,complete response letter)。
平心而論,FDA對於信迪利單抗的評估還算公允,ORIENT-11試驗的主要終點,對照組選擇,研究人羣,以及知情同意程序等方面確實與FDA的要求存在差距,而且沒有在臨牀試驗前就臨牀設計與FDA溝通達成一致。
雖然信迪利單抗很有可能在種族之間沒有差別,療效理論上可以轉化到美國人羣,但是沒有直接的臨牀數據證明這一點。最關鍵是FDA在轉移性非小細胞肺癌一線治療這個適應症上已經批准了10個PD-1/PD-L1單藥或聯合用藥產品,信迪利單抗不能解決未滿足的臨牀需求。應該説FDA的這個審評原則與中國CDE倡導的以臨牀價值為導向的精神是完全一致的,都是鼓勵更新或更好、能夠解決未滿足臨牀需求的創新葯研發。
所以這個ODAC會議結果並不代表FDA完全關上了新葯產品用中國臨牀數據在美國上市的這個大門,FDA在會議上明確表示只要滿足FDA對於海外數據的接受要求,針對未滿足的臨牀需求(unmet medical needs),罕見病(MRCT難以開展)、全創新葯等3種情況還是會給予相應的監管靈活性,比如鼻咽癌(NPC,Nasopharyngeal Carcinoma)和兒童癌症,在美國屬於罕見病,也沒有已經批准的有效治療,那麼還是存在用中國的臨牀數據獲得美國批准上市的可能性。
在ODAC委員會投票闡明瞭立場之后,現在在信達面前好像只有一條可行的路徑,那就是與已經上市的PD-1產品(比如K藥)用OS做終點做一個頭對頭的非劣效MRCT試驗來證明信迪利單抗在美國病人中的療效不劣於K藥。但是這樣的開發路徑不僅投資高,時間長,在美國招募病人會有很大挑戰,而且還具有非常大的不確定性,所以感覺信達/禮來可能不會採取這樣的路徑。
其實信達/禮來可能是基於商業上的考慮,選擇了非小細胞肺癌一線治療這樣一個在美國已經有很多PD-1產品獲批上市、門檻非常高的適應症申報BLA,但是從ODAC會議的結果來看,這種選擇還是值得商榷的。
那麼是否有其他的路徑?我覺得在ODAC會議上,FDA其實反覆強調對於未滿足的臨牀需求還是會給予相應的監管靈活性,所以更為可行的一條路徑應該是像君實和康方申請鼻咽癌的適應症一樣,找到一個罕見病、具有未滿足的臨牀需求、而且沒有已經批准的PD-1產品的適應症來獲得上市批准,然后通過降低價格和擴大標籤使用來獲得更多的市場份額。
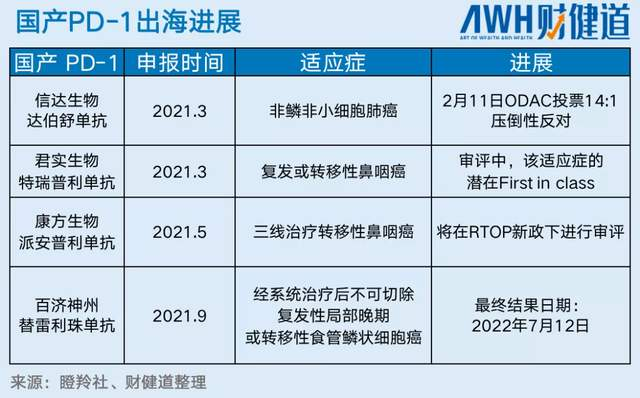
而在信達的信迪利單抗之后,還有3款國產PD-1均已於去年提交美國BLA申請,今年等着排隊上市了,分別是君實生物的特瑞普利單抗,康方生物的派安普利單抗和百濟神州替雷利珠單抗。替雷利珠單抗擁有全球多中心MRCT試驗數據,而特瑞普利單抗和派安普利單抗則是瞄準了鼻咽癌的適應症,理論上能夠滿足FDA的准入要求或者獲得監管靈活性,相信2022年肯定會聽到中國的PD-1產品在美國獲批上市的好消息。
五、結束語
FDA通過這次ODAC會議給中國生物製藥企業上的這堂公開課可能最大的作用就是明確給出了中國新葯到美國上市的准入要求,讓許多正在用中國數據申報BLA的中國公司的美國夢可能隨之破滅,但是也給認真做藥具備實力出海的新葯公司提供了一個明確的路徑:
從臨牀1期開始就做好國際註冊策略和臨牀開發計劃,構建一個包括醫學,臨牀運營,臨牀藥理,數據管理、生物統計、臨牀安全和藥物警戒,和註冊在內的跨部門團隊,進一步熟悉國際監管要求,通過與國際監管機構的溝通和合作,開展MRCT國際多中心臨牀試驗,開發出真正能夠解決中國和歐美未被滿足臨牀需求的新葯。
FDA在ODAC會議前也提出希望把中國納入FDA發起並主導的多國同步平行審評計劃Project Orbis,這樣可以促進MRCT的發展,加快創新腫瘤藥在全球主要市場同步上市的速度,當然這對於中國的本土創新企業提出了更高的要求,畢竟在歐美藥企的研發經驗和水平還是要高於中國藥企的情況下,全球加速上市也意味着對於中國藥企的更大挑戰。
但是國際化是中國創新葯發展的必然路徑,預計本次事件將進一步刺激中國創新葯企加快出海蔘與全球競爭的步伐,未雨綢繆早日構築國際競爭優勢,提升國產創新葯研發實力。
信達/禮來這次與FDA的ODAC會議長期來看肯定會對中國生物製藥公司的國際化和成功出海起到促進的作用,成為中國創新葯發展歷史上的一個重要里程碑。整個過程可以用北京2022年冬奧會上,羽生結弦在男子單人自由滑中未能成功挑戰阿克塞爾四周跳摔倒之后,央視解説員陳瀅為他賦詩中寫的這段話來形容:「守一座守不住的城,打一場打不贏的仗,天意終究難參,假若登頂成憾,與君共添青史幾篆,成敗也當笑看。」
特別感謝:羅氏基因泰克公司全球註冊高級總監應嘉博士,啟德醫藥生物統計副總裁夏毅博士,安博生物法規事務副總裁洪筱玲博士,Relay Therapeutics法規事務副總裁黃桂林,埃格林醫藥CEO杜新博士在百忙之中對本文進行了認真審閲,給予了寶貴意見。
參考文獻
[1] Beaver, J. A., & Pazdur, R. "Dangling" Accelerated Approvals in Oncology, N Eng J Med. 2021 May 6.
[2] Beaver, J. A., & Pazdur, R. The Wild West of Checkpoint Inhibitor Development. N Engl J Med. 2021 Dec 15.
[3] Singh, H., & Pazdur, R. Importing oncology trials from China: a bridge over troubled waters? Lancet Oncol. 2022 Feb 4
[4] Steven Lemery , & Richard Pazdur, Approvals in 2021: dangling Accelerated Approvals, drug dosing, new approvals and beyond, Nat Rev Clin Oncol. 2022 Feb 8;1-2.
[5] Adam Feuerstein, ‘I have a right to change my mind’: A top FDA regulator is unapologetic over his about-face on Chinese cancer drugs, STAT Feb. 8, 2022
本文來自微信公眾號:同寫意(ID:tongxieyi),作者:萬里 博士(啟德醫藥高級副總裁和國際註冊事務部負責人、美中藥協註冊社羣負責人)
推薦文章
提價83%需求卻暴增400%!智譜、MiniMax鎖定大模型定價權,AI Agent元年即將開啟?

港股見底了嗎?北水大舉加倉逾600億港元!大行看好4月做多窗口來臨,十大金股一圖睇全
華盛早報 | 伊朗與阿曼擬共管霍爾木茲!美股V型反轉;伊朗襲擊甲骨文、亞馬遜數據中心;港美股今日因假期休市一天
諾和諾德稱:口服版Wegovy減肥效果優於禮來GLP‑1口服藥
美股機會日報 | 特朗普粉碎停戰幻想!恐慌指數飆升12%,納指期貨跌約2%;美油期貨暴漲超9%!油氣股飆升,美國原油基金ETF漲超9%
清明休市提醒 | 港股本周五休市,下周三恢復交易;美股周五休市一天
油價上漲 此前特朗普表示伊朗衝突可能在未來幾周升級
野村:特朗普講話未能發出局勢降温的明確信號
